
Hueckel Molecular Orbital HMO
4,2star
116 recensioner
10 tn+
Nedladdningar
Ingen åldersgräns
info



Om appen
Hueckel molekylorbitalteori - HMO
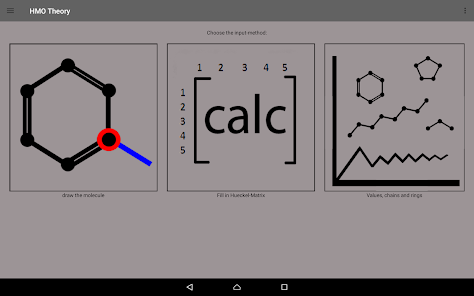
Denna app beräknar molekyler genom Hueckel-ungefärlig.
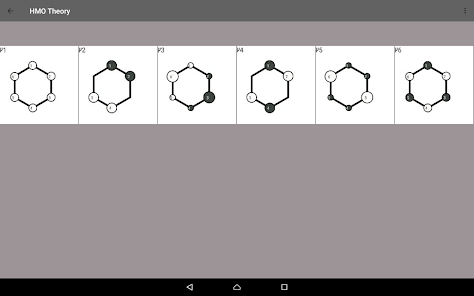
Du kan antingen in direkt av topologin matrisen eller genom de genomförda egen ritning systemet.
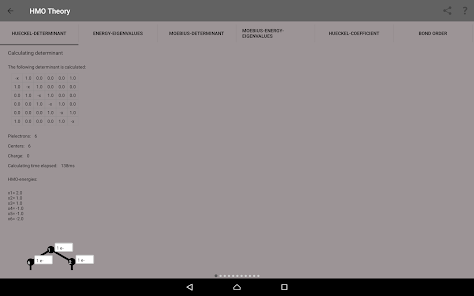
Detta värden kommer att beräknas genom att lösa en avgörande faktor för den angivna matrisen:
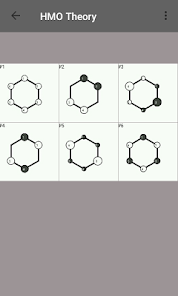
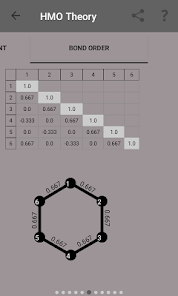
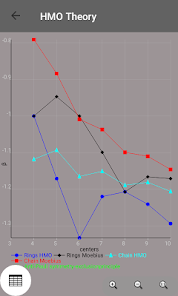
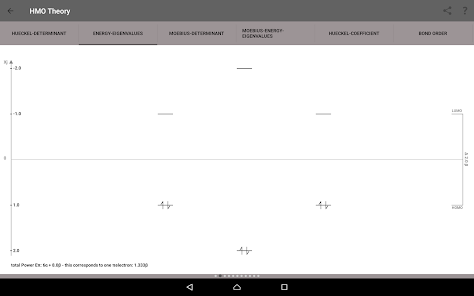
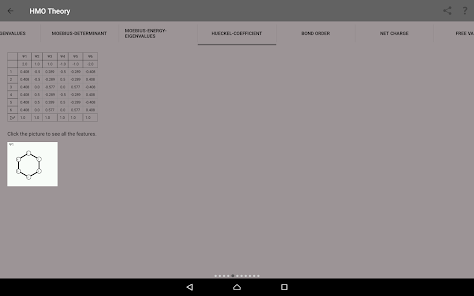
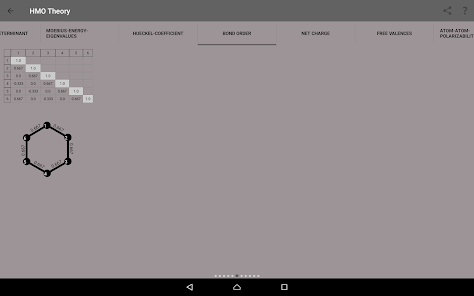
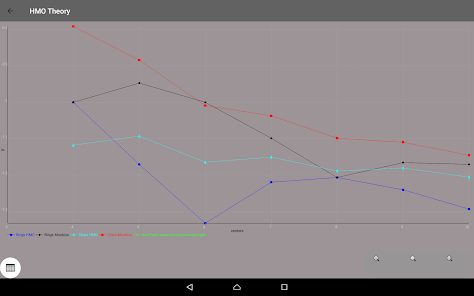
Egenvärden, energinivådiagram, bond beställning, laddning beställning, nettoladdning, fria valenser, atom-atom / bond-atom / bond-bond polariserbarhet.
Alla värden är grafiskt bearbetas och kan exporteras till PDF.
Du kan beräkna fysiska kvantmekaniska effekter på din egen mobiltelefon eller Tablet.
Perfekt för utbildning i skolan och universitetet.
Utvecklat vid Tekniska universitetet i Darmstadt - Teoretisk Fysikalisk kemi / TU Darmstadt
Om du är intresserad av att hjälpa till att översätta denna app? Har du några förslag till förbättringar eller hittat några misstag ?. Vänligen kontakta p.giel@gmx.de
Denna app beräknar molekyler genom Hueckel-ungefärlig.
Du kan antingen in direkt av topologin matrisen eller genom de genomförda egen ritning systemet.
Detta värden kommer att beräknas genom att lösa en avgörande faktor för den angivna matrisen:
Egenvärden, energinivådiagram, bond beställning, laddning beställning, nettoladdning, fria valenser, atom-atom / bond-atom / bond-bond polariserbarhet.
Alla värden är grafiskt bearbetas och kan exporteras till PDF.
Du kan beräkna fysiska kvantmekaniska effekter på din egen mobiltelefon eller Tablet.
Perfekt för utbildning i skolan och universitetet.
Utvecklat vid Tekniska universitetet i Darmstadt - Teoretisk Fysikalisk kemi / TU Darmstadt
Om du är intresserad av att hjälpa till att översätta denna app? Har du några förslag till förbättringar eller hittat några misstag ?. Vänligen kontakta p.giel@gmx.de
Uppdaterades den
Säkerhet börjar med förståelsen av hur utvecklare samlar in och delar din data. Praxis för dataintegritet och säkerhet varierar beroende på användning, region och ålder. Utvecklaren har tillhandahållit denna information och kan uppdatera den med tiden.


Betyg och recensioner
4,2
107 recensioner
Nyheter
! Code-Rev
* Add Feedback Mailgun
* Remove ACRA, implement Firebase
* Remove Bugs
* Update SDK
* Update Android SupportLibs
* Add Feedback Mailgun
* Remove ACRA, implement Firebase
* Remove Bugs
* Update SDK
* Update Android SupportLibs
Appsupport
Om utvecklaren
PATRICK GIEL
P.giel@gmx.de
Seligenstädter Str. 14
63500 Seligenstadt
Germany
undefined